
|
|
November 18, 2025: Release of ENDscript v2.2 integrating the AlphaFold Protein Structure Database (with around one million entries from the ‘Model Organism Proteomes’, ‘Global Health Proteomes’ and ‘Swiss-Prot’ subsets). October 25, 2025: We are pleased to announce the release of our new web server: NESPript  It has been designed for the representation of multiple alignments of nucleic acid sequences, including the visualization of their secondary structures. |
| What is ENDscript? |
|---|
|
ENDscript is a user-friendly web server that extracts and renders a comprehensive analysis of primary to quaternary protein structure information in an automated way.
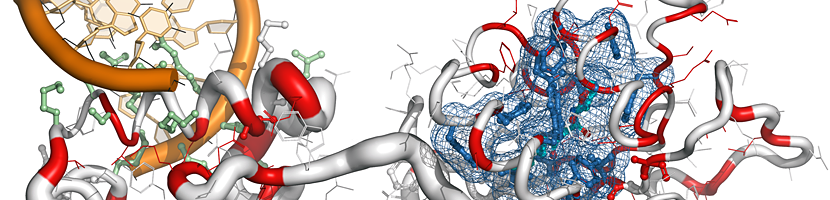

Nice overview of ENDscript and related programs by Dr. Brianna Bibel. For more engaging content on structural biology and biochemistry, visit her Bumbling Biochemist YouTube channel. |
| ENDscript key points |
|---|
|
From a typed PDB identifier or an uploaded PDB/CIF file, ENDscript quickly generates the following downloadable illustrations:
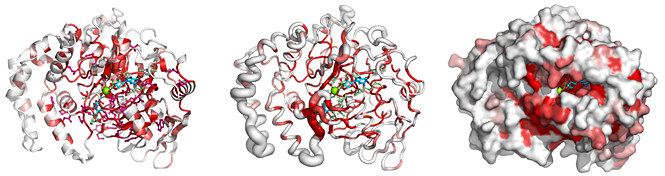

|
| ENDscript is easy! |
|---|
|
We have put a lot of effort into making ENDscript fast and easy to use:
|
| How does ENDscript work? | |
|---|---|
ENDscript taked as a query either a four-digit PDB identifier (e.g. 2CAH) or an uploaded coordinate file in PDB or CIF format (NMR and crystallographic structures are supported).ENDscript’s automated pipeline involves several sequence and structure analysis software and is divided into three sequential phases:
|
ENDscript can handle up to 3,000 distinct sequences adorned with their secondary structure elements and render their representation in the gigantic Tapestryformat (0.8 × 3.3 meters)! 
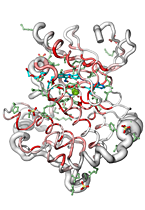
|
| Examples of ENDscript outputs | |||||
|---|---|---|---|---|---|
|
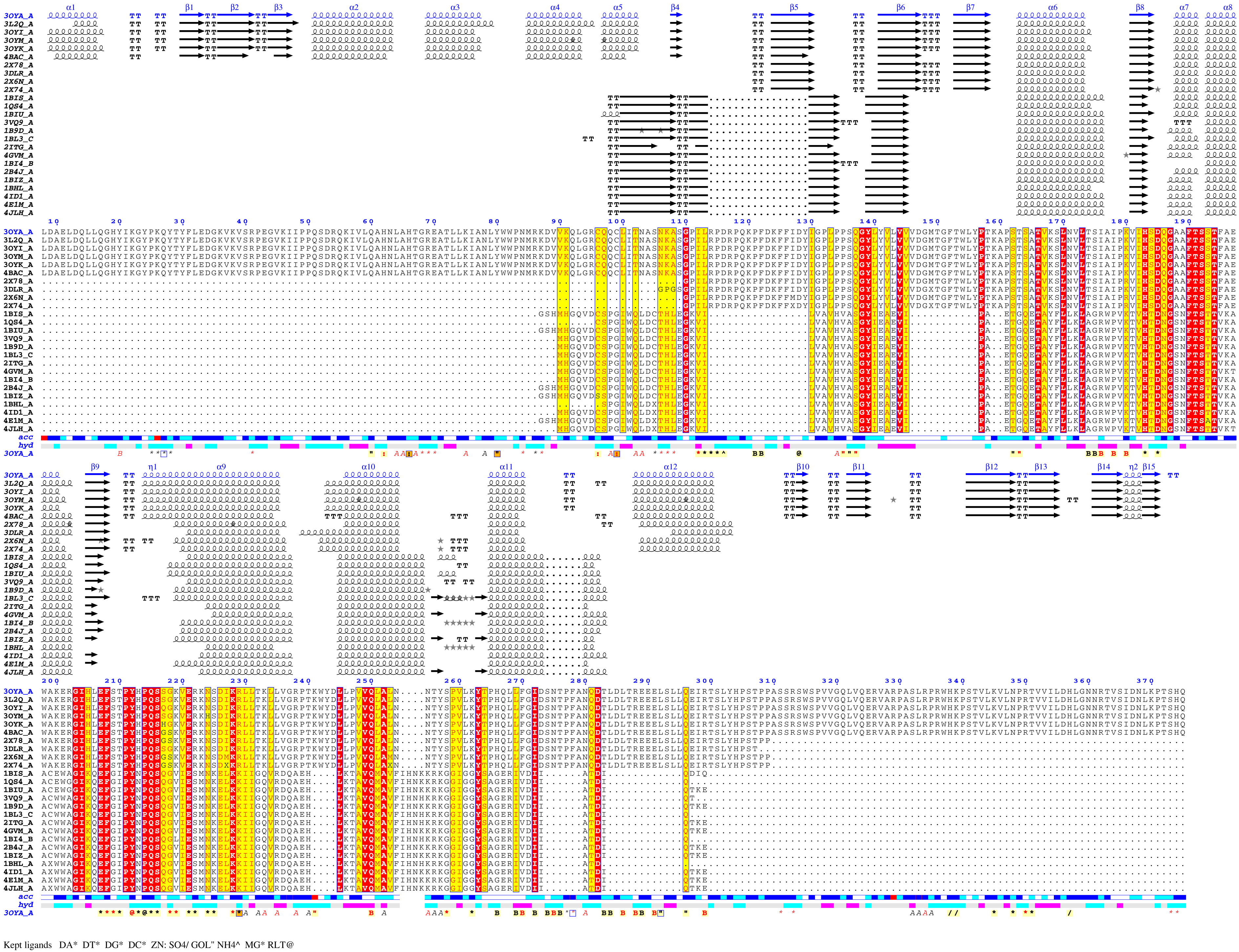
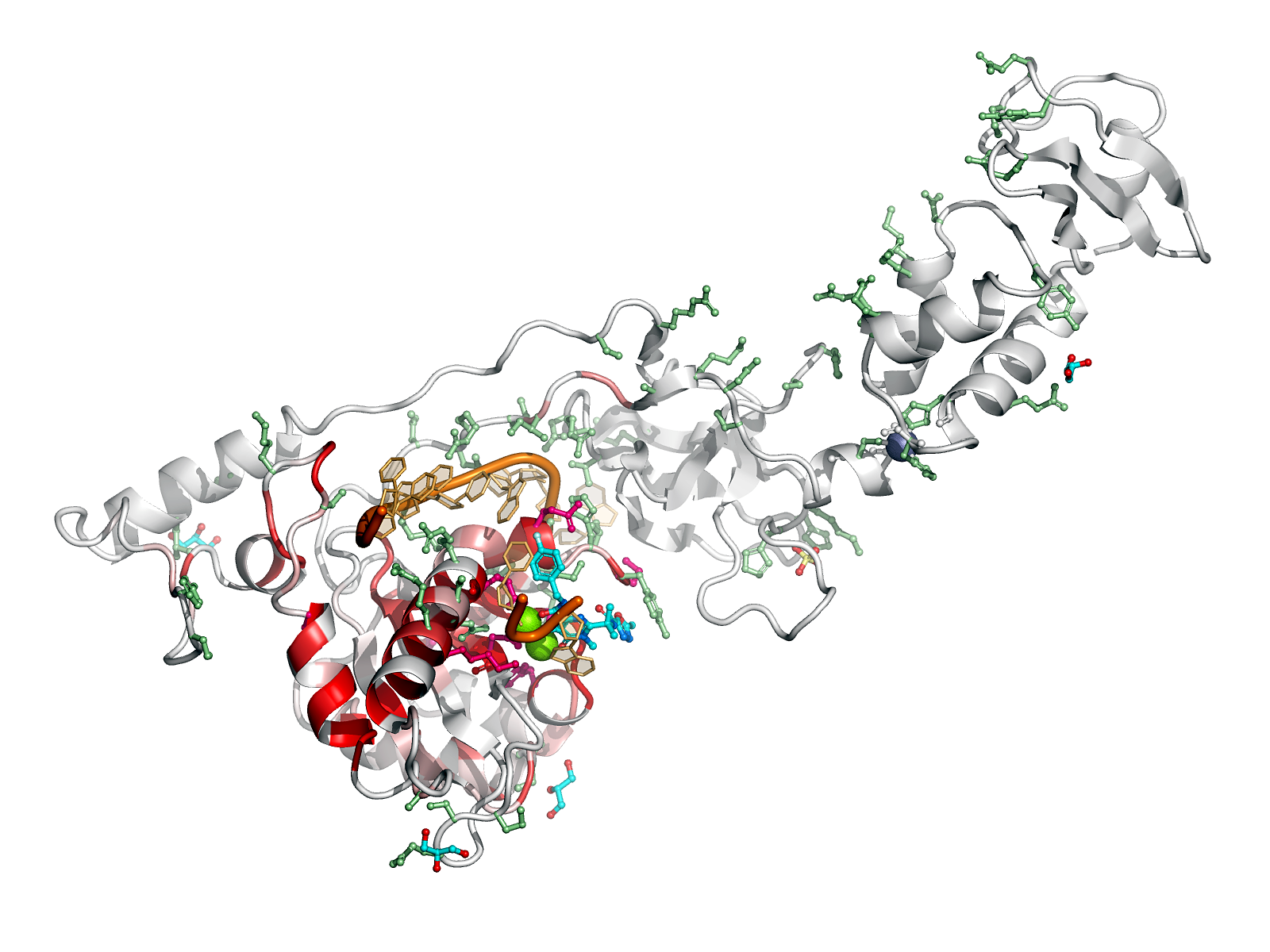
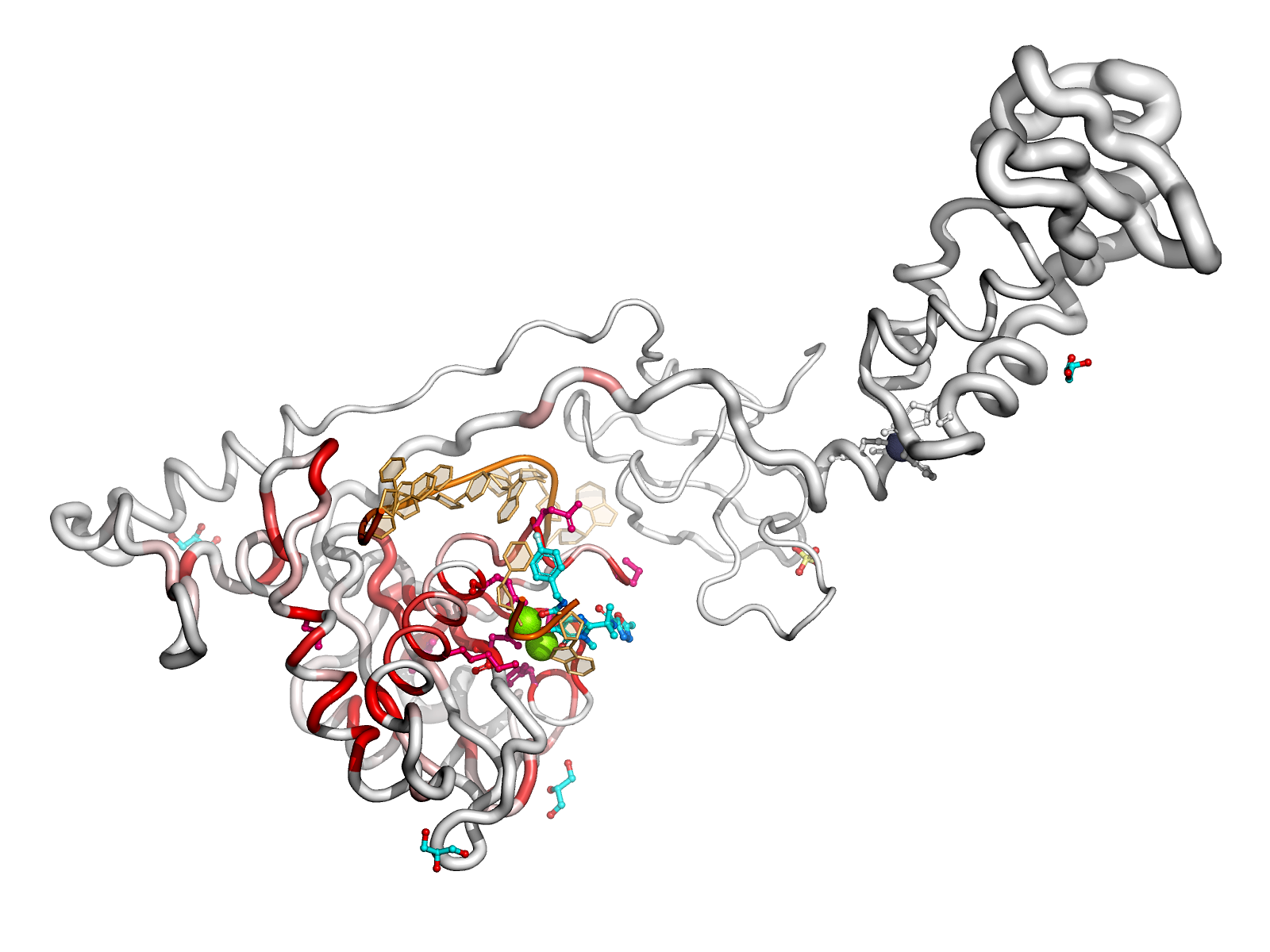
Here are some excerpts from ENDscript-generated flat figures and PyMOL sessions (PDB entry 3OYA). (Click on the thumbnails to access full-size pictures) | |||||
 ENDscript phase 1 Query only |
 ENDscript phase 2 Query and homologous |
 ENDscript phase 3 PyMOL Cartoon representation |
 ENDscript phase 3 PyMOL Sausage representation |
 ENDscript phase 3 PyMOL Surface representation |
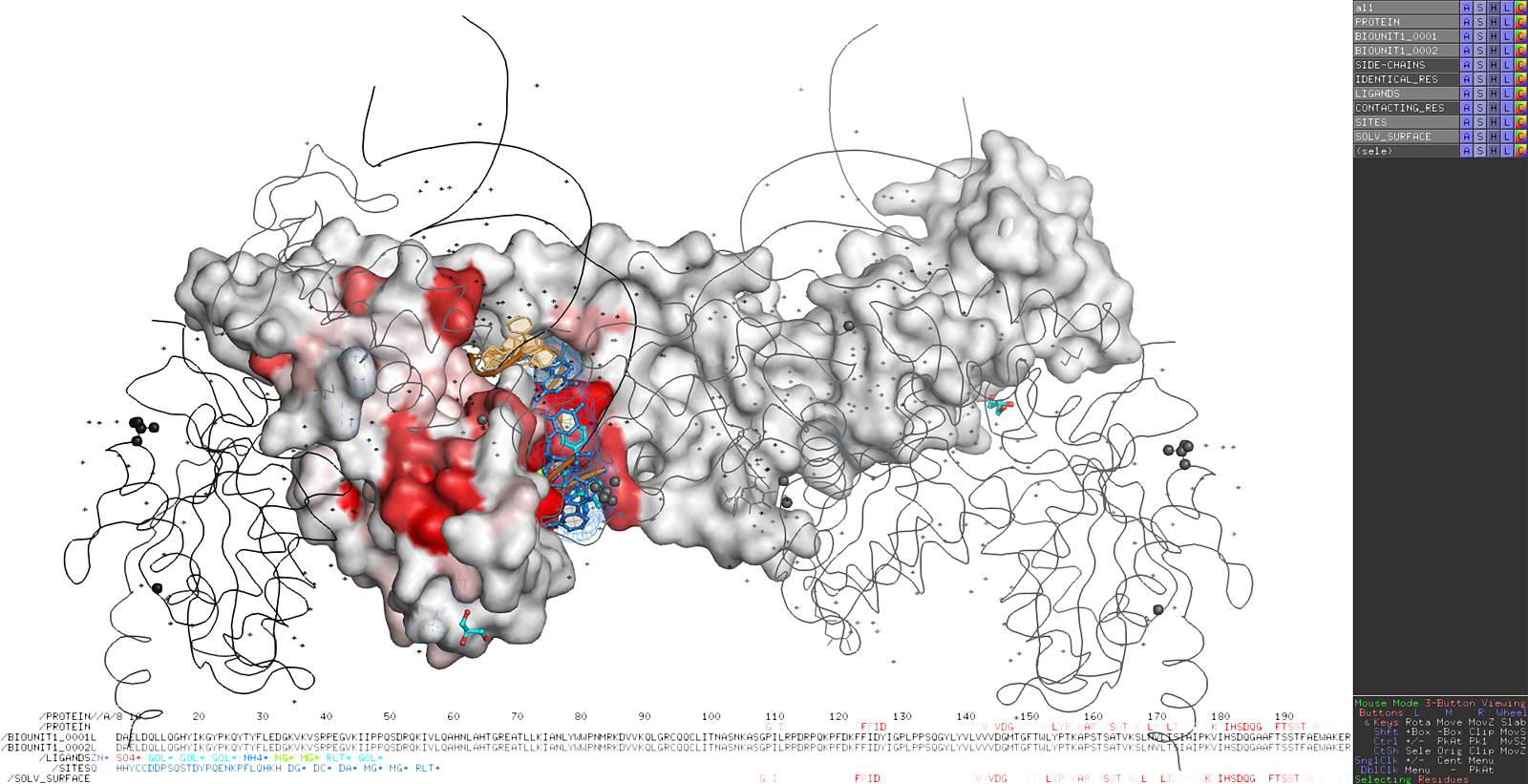 ENDscript phase 3 PyMOL screen capture |
| |||||
| ENDscript is open to external bioinformatics services |
|---|
|
We have designed ENDscript 2 as an open platform for the visualization of multiple biochemical and structural information:
|
| Terms of use |
|---|
|
|
|
© 2005-2026 The ENDscript authors & CNRS - Contact: espript@ibcp.fr ESPript is an SBGrid supported application |